


Conheça as principais etapas para autorização temporária de uso emergencial das vacinas.
A natureza não dá saltos (Natura non facit saltus). A frase do botânico e médico sueco Carlos Lineu sintetiza a noção de que as mudanças na ciência ocorrem de forma gradativa. A ideia ajuda a explicar o caminho para o desenvolvimento das vacinas e os passos que precisam ser dados até que elas cheguem até as salas de imunização em todo o país.
Para que a sociedade acompanhe, com total transparência, as etapas para autorização do uso emergencial das vacinas, que vêm exigindo tempo, investimento e, sobretudo, muito esforço humano, a Anvisa elaborou infográficos e roteiros explicativos com links para as matérias já publicadas. Confira.
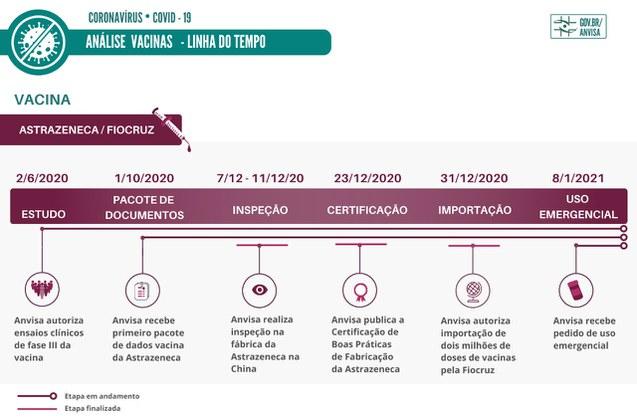
Vacina AstraZeneca/Fiocruz
- A Anvisa autorizou, em 2/6/20, os primeiros ensaios clínicos da fase III da vacina.
Quatro meses depois, em 1º de outubro, a Agência recebeu o primeiro pacote de dados.
Em dezembro, inspetores da Anvisa iniciam a inspeção na fábrica da AstraZeneca, na China. Foram avaliados pontos como o Sistema de Gestão da Qualidade farmacêutica da empresa, Gerenciamento de Risco, Gerenciamento de Documentos e Plano Mestre de Validação. No segundo dia de inspeção, em 8/12, a equipe verificou, entre outros aspectos, as instalações produtivas destinadas à produção do insumo farmacêutico ativo (IFA). A atividade de inspeção na fábrica é encerrada em 11/12.
A Certificação de Boas Práticas de Fabricação (CBPF) da AstraZeneca é publicada em 23/12. O documento é um dos requisitos da análise do registro da vacina.
A importação excepcional de dois milhões de doses de vacinas pela Fiocruz foi aprovada em 31 de dezembro, mesmo dia em que o pedido de importação foi protocolado pela Fiocruz. A autorização teve como objetivo antecipar a disponibilização de vacinas até que o produto seja aprovado.
No dia 8 de janeiro, a Fiocruz deu entrada na Anvisa ao pedido de autorização temporária de uso emergencial para os dois milhões de doses de vacinas desenvolvidas pela empresa AstraZeneca e que serão importadas do laboratório Serum, sediado na Índia. Nesse mesmo dia, tem início a triagem dos documentos encaminhados juntamente com o pedido.
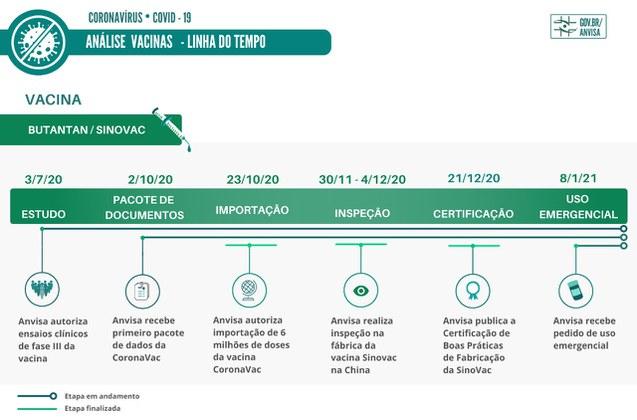
Vacina Butantan/Sinovac
A Anvisa autorizou, em 3/7/20, os primeiros ensaios clínicos da fase III da vacina.
Três meses depois, em 2/10, a Anvisa recebeu o primeiro pacote de dados.
Foi aprovada, em 23/10, a importação de seis milhões de doses da vacina CoronaVac. A liberação permitiu a internalização da vacina no Brasil. O produto deve ficar sob a guarda da empresa responsável pelo registro no país até que seja autorizado o uso.
Inspetores da Anvisa iniciam, em 30/11, o processo de inspeção na empresa Sinovac. Foram avaliados pontos como o Sistema de Gestão da Qualidade farmacêutica da empresa, Gerenciamento de Risco, Gerenciamento de Documentos e Plano Mestre de Validação. No segundo dia de inspeção, a equipe se concentrou na verificação das Boas Práticas de Fabricação da vacina CoronaVac. O processo de inspeção é encerrado no dia 4 de dezembro.
A Certificação de Boas Práticas de Fabricação (CBPF) da Sinovac é publicada em 21/12. Essa etapa é um dos pré-requisitos para a continuidade do processo de registro da vacina e do pedido de autorização de uso emergencial.
No dia 8 de janeiro, a Anvisa recebeu pedido de autorização temporária de uso emergencial do Butantan. As primeiras 24 horas foram utilizadas para triagem do processo e verificação dos documentos.
No dia 17 de janeiro de 2021, um dia antes de encerrar o prazo para a análise dos pedidos de uso emergencial, a Anvisa vai decidir em uma reunião extraordinária da Diretoria Colegiada se aprova ou não os pedidos. Depois de protocolados os processos, a Agência teve 10 dias para analisá-los.
Com toda essa evolução, o dia histórico de hoje, 17 de janeiro de 2021, temos a aprovação, mesmo que com ressalvas, das duas vacinas para iniciar a vacinaÇão no Brasil.